Le séquençage à haut débit permet aujourd’hui de séquencer la majorité des acides nucléiques (ADN, ARN) présents dans un échantillon d’eau de mer et d’en caractériser les communautés planctoniques, composées principalement d’organismes microscopiques. Ces jeux de données dites métagénomiques atteignent désormais l’ordre du téra1 , et offrent l’opportunité d’étudier l’impact des conditions environnementales sur la composition de ces communautés jouant un rôle clef dans la régulation du climat global. À partir de données initialement collectées dans le cadre de l’expédition Tara Oceans, une équipe de recherche2 de Sorbonne Université, du CNRS et du Muséum national d’Histoire naturelle a combiné les approches de métagénomique à haut-débit à des techniques d’apprentissage automatique issues du machine learning. L’objectif : prédire la composition génomique du plancton à partir du contexte environnemental pour, à terme, pouvoir prédire les effets du changement climatique sur ces micro-organismes. Les résultats de leur étude* ont été publiés le 16 juillet 2021 dans la revue Nature Communications.
Un défi majeur du XXIème siècle est de mieux comprendre et prédire les effets du changement climatique et des actions de l’homme sur le fonctionnement des écosystèmes. Les micro-organismes marins jouent un rôle crucial dans la régulation du climat de la planète, les cycles biogéochimiques globaux et les réseaux trophiques océaniques. Ainsi le plancton des océans produit la moitié de l’oxygène que nous respirons et est à la base des réseaux trophiques alimentant les pêcheries. Une énorme quantité de données issues de séquençage à haut-débit ont été générées ces dix dernières années sur les communautés planctoniques naturelles, de l’ordre du téra, notamment grâce à la multiplication des expéditions océanographiques de grande échelle, permettant d’estimer, de plus en plus précisément, leur étonnante diversité. Face à cette quantité de données métagénomiques sans précédent, il apparaît nécessaire de développer des méthodologies innovantes fondées sur les données pour quantifier et prédire le rôle écologique de ces séquences d’ADN et d’ARN. L’enjeu est donc de parvenir à intégrer l’ensemble de ces séquences, dont une grande partie ont des rôles encore inconnus, dans un cadre statistique permettant de lier leur présence et leur abondance à des données environnementales hétérogènes, en limitant au maximum les choix a priori de fonctions ou d’organismes d’intérêt. Cela permettrait de mieux comprendre les mécanismes clefs impliquant les micro-organismes marins dans le fonctionnement global des océans, ainsi que leur dépendance aux conditions environnementales. On pourrait alors mieux mesurer l’impact potentiel du changement climatique sur les écosystèmes marins.
Un défi majeur du XXIème siècle est de mieux comprendre et prédire les effets du changement climatique et des actions de l’homme sur le fonctionnement des écosystèmes. Les micro-organismes marins jouent un rôle crucial dans la régulation du climat de la planète, les cycles biogéochimiques globaux et les réseaux trophiques océaniques. Ainsi le plancton des océans produit la moitié de l’oxygène que nous respirons et est à la base des réseaux trophiques alimentant les pêcheries. Une énorme quantité de données issues de séquençage à haut-débit ont été générées ces dix dernières années sur les communautés planctoniques naturelles, de l’ordre du téra, notamment grâce à la multiplication des expéditions océanographiques de grande échelle, permettant d’estimer, de plus en plus précisément, leur étonnante diversité. Face à cette quantité de données métagénomiques sans précédent, il apparaît nécessaire de développer des méthodologies innovantes fondées sur les données pour quantifier et prédire le rôle écologique de ces séquences d’ADN et d’ARN. L’enjeu est donc de parvenir à intégrer l’ensemble de ces séquences, dont une grande partie ont des rôles encore inconnus, dans un cadre statistique permettant de lier leur présence et leur abondance à des données environnementales hétérogènes, en limitant au maximum les choix a priori de fonctions ou d’organismes d’intérêt. Cela permettrait de mieux comprendre les mécanismes clefs impliquant les micro-organismes marins dans le fonctionnement global des océans, ainsi que leur dépendance aux conditions environnementales. On pourrait alors mieux mesurer l’impact potentiel du changement climatique sur les écosystèmes marins.
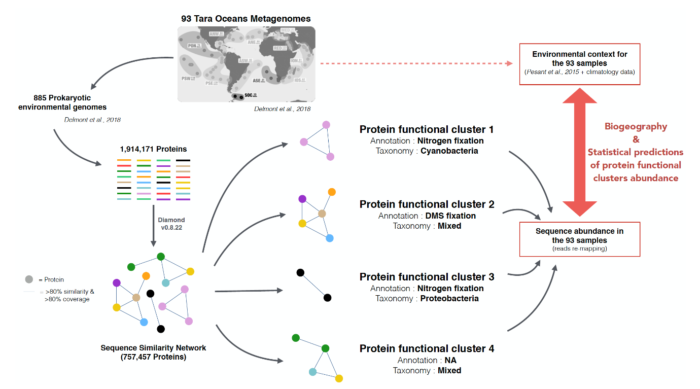
Résumé graphique des méthodes employées par Faure et al. (2021). Près de 2 millions de protéines provenant de génomes de bactéries et d’archées marines sont distribuées dans plus de 200 000 familles protéiques via un réseau de similarité de séquence. Les abondances de ces familles protéiques sont ensuite qualifiées au sein de 93 échantillons afin d’étudier le lien ente leur abondance et les conditions environnementales dans l’océan global.
Applicable à tout jeu de données de métagénomique, et tout type d’écosystème (marin, eau douce, sol, microbiote, etc.), cette approche constitue une première étape vers des prédictions quantitatives de la composition fonctionnelle des communautés de micro-organismes à partir de paramètres environnementaux. Ces résultats pourraient ouvrir la voie à des prédictions “données-centrées” des effets potentiels du changement climatique sur les communautés de microorganismes, ainsi que sur leurs rôles au sein des écosystèmes.
1 Soit 10^12 paires de bases nucléotidiques.
2 Laboratoire d’océanographie de Villefranche (LOV, Sorbonne Université/CNRS), Institut de systémique, évolution, biodiversité (ISYEB, Sorbonne Université/CNRS/MNHN/EPHE-PSL), Institut des sciences du calcul et des données (ISCD) de Sorbonne Université
*Ces travaux ont été en partie financés par une bourse de thèse du programme doctoral Interfaces Pour le Vivant (IPV) et par l’Institut des Sciences du Calcul et des Données (ISCD) de Sorbonne Université.
Méthodologie
Dans cette étude, les scientifiques proposent une approche basée sur l’analyse de réseaux de similarité de séquences, appliquée à 885 génomes de bactéries et d’archées marines précédemment obtenus par assemblage de métagénomes du projet Tara Oceans. Cette approche leur a permis une comparaison exhaustive de 757 457 séquences, ensuite rassemblées en 233 756 familles protéiques. Parmi ces familles, 15% étaient fonctionnellement non annotées, c’est-à-dire que leur rôle était totalement inconnu. Ils ont étudié la distribution de ces familles protéiques dans l’océan à l’échelle globale. Grâce à des méthodes d’apprentissage automatique (machine learning) utilisant comme prédicteurs un ensemble de paramètres physico-chimiques caractérisant les conditions environnementales des lieux d’échantillonnage, ils ont pu prédire l’abondance de 14 585 familles protéiques, dont 1 347 fonctionnellement non annotées. Ils ont identifié les provinces biogéographiques, c’est-à-dire les grandes régions de l’océan, comme étant les meilleurs prédicteurs de l’abondance des familles protéiques, et ont démontré que la Mer Méditerranée et l’Océan Austral constituaient des points particulièrement originaux en termes de composition en familles protéiques.
Résumé graphique des méthodes employées par Faure et al. (2021). Près de 2 millions de protéines provenant de génomes de bactéries et d’archées marines sont distribuées dans plus de 200 000 familles protéiques via un réseau de similarité de séquence. Les abondances de ces familles protéiques sont ensuite qualifiées au sein de 93 échantillons afin d’étudier le lien ente leur abondance et les conditions environnementales dans l’océan global.
Référence :
Towards omics-based predictions of planktonic functional composition from environmental data, Faure Emile, Ayata Sakina-Dorothée, Bittner Lucie, Nature Communications, July 16th 2021.
doi :10.1038/s41467-021-24547-1
À propos de Sorbonne Université :
Sorbonne Université, née de la fusion des universités Paris-Sorbonne et Pierre et Marie Curie, est une université pluridisciplinaire de recherche intensive de rang mondial. Sorbonne Université couvre tout l’éventail disciplinaire des lettres, de la médecine et des sciences. Ancrée au cœur de Paris, présente en région, elle est engagée pour la réussite de ses étudiants et s’attache à répondre aux enjeux scientifiques du 21e siècle et à transmettre les connaissances issues de ses laboratoires et de ses équipes de recherche à la société toute entière. Grâce à ses près de 55 000 étudiants, 6 700 enseignants-chercheurs et chercheurs et 4 900 personnels administratifs et techniques qui la font vivre au quotidien, Sorbonne Université se veut diverse, créatrice, innovante et ouverte sur le monde. Avec le Museum National d’Histoire Naturelle, l’Université de Technologie de Compiègne, l’INSEAD, le Pôle Supérieur Paris Boulogne Billancourt et France Education International, elle forme l’Alliance Sorbonne Université. La diversité des membres de l’Alliance Sorbonne Université favorise une approche globale de l’enseignement et de la recherche. Elle promeut l’accès de tous au savoir et développe de nombreux programmes et projets communs en formation initiale, continue et tout au long de la vie dans toutes les disciplines. Sorbonne Université est membre de l’Alliance 4EU+, un nouveau modèle d’université européenne, avec les universités Charles de Prague (République Tchèque), de Heidelberg (Allemagne), de Varsovie (Pologne), de Milan (Italie) et
de Copenhague (Danemark). www.sorbonne-universite.fr @ServicePresseS